Browsing entries in the InterPro website¶
You can get to entry pages in InterPro in lots of different ways. Commonly this will involve clicking on a link to an entry from one of the search methods. This section describes the different types of entries and what you will find for each of their pages.
There are 8 categories of pages in InterPro:
The following entry data tabs are available when appropriate. We describe each in detail in the first entry page it appears in. Most entry data tabs will be described within the InterPro entry page.
InterPro entry page¶
An InterPro entry represents a unique protein homologous superfamily, family, domain, repeat or important site based on one or more signatures provided by the InterPro member databases.
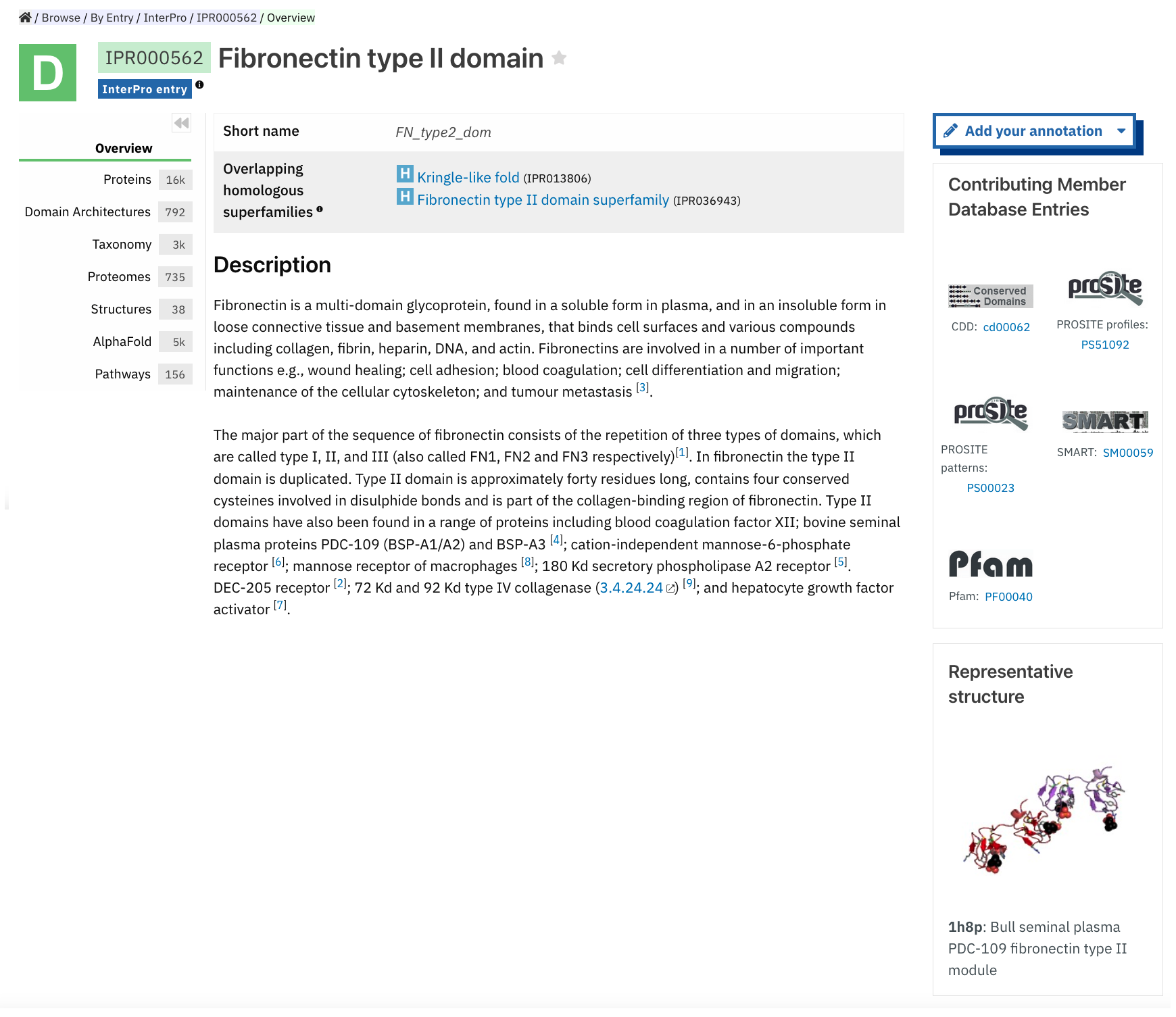
InterPro entry pages give a brief description of the entry, name and unique InterPro accession. The InterPro entry type (homologous superfamily, family, domain, repeat or site) is also indicated by an icon (e.g. a D with a green background for a domain).
Clicking on the star symbol next to the entry name will save the entry as a Favourite. The full list of saved entries is available in the Favourites Entries component in the homepage. More information about the data provided in an
On the right hand side, the Provide feedback button links to a Feedback page, which allows the user to suggest updates to the InterPro entry annotation. Below, the Contributing Member Database Entry/ies integrated into the InterPro entry are listed with links to the corresponding member database pages. At the bottom of this column, if any experimentally solved structure is available, a Representative structure shows a small static 3D representation, the corresponding PDB ID and name, and a link to the structure entry page. The chosen representative structure is picked from structures that match the entry and have a resolution of less than 2 Angstroms. In this refined dataset, the representative structure is identified as the one exhibiting the highest coverage ratio for the entry, where a minimum of 50% of the residues in the structure are covered by the entry.
Overlapping homologous superfamilies and/or Relationships to other entries are indicated where available.
InterPro entry page can be found in the InterPro Entries: essential information section of the documentation.
Additional tabs in the left-hand side menu provide further information about the entry, and are displayed when the data is available. Types of data that may be available in the menu of an InterPro entry page include: Proteins, Domain architectures, Taxonomy, Proteomes, Structures, AlphaFold, Pathways and Interactions.
Although most InterPro entries remain carefully reviewed by our curators, some type Family entries containing signatures from PANTHER, NCBIfam or CATH-Gene3D which cover approximately the whole protein length are AI-generated. For these entries, the name, short-name and description have been generated automatically
using a Large Language Model. All AI-generated content is flagged as such with an  tag. Please note that this content may not have been subjected to
curator review when interpreting related results. When the content has been reviewed the
status tag is updated accordingly. The possible status are:
tag. Please note that this content may not have been subjected to
curator review when interpreting related results. When the content has been reviewed the
status tag is updated accordingly. The possible status are:
AI-generated Unreviewed (default): the entry hasn’t been verified by a curator
AI-generated Reviewed and updated: the entry has been verified and sligthly updated by a curator (e.g. literature references have been added and grammar, style and other minor issues corrected)
The AI status is highlighted on the entry page in the tooltip when hovering over the tag next to the entry name and short name,
and with the  or
or  tags above the entry descriptions.
tags above the entry descriptions.
If the AI-generated the entry has been verified and significantly updated by a curator, it will be marked as curated and the tag will no longer be visible.
More information on AI-generated content can be found in the AI-generated content section.

InterPro AI-generated entry page for IPR051632. Name, short-name and description have been generated using a Large Language Model and are flaggged accordingly.¶
Proteins¶
List of proteins that are included in this entry displayed in a table. There is an option to display only proteins that have been manually curated in UniprotKB (reviewed), only proteins that have been automatically annotated (unreviewed), or all proteins (both, default).
For each protein, the table displays the UniProt ID, name, corresponding gene, the organism where it is found, a link to the predicted structure in AlphaFold structure prediction page, which now includes predictions from the Big Fantastic Virus Database (BFVD), and a small protein viewer that highlights the region of the protein matched by the InterPro entry.
Domain architectures¶
Provides information about the different domains arrangements for the proteins matching this entry based on Pfam signatures. For InterPro entries, it provides information about where the domain is located in protein sequences and what, if any, combinations arise with other domains. Domain architectures can be downloaded in JSON and TSV formats through the Export button.
Taxonomy¶
List of species this entry is matching, based on data from UniProt taxonomy. The information can be displayed in 4 different ways through the view options menu:
Table with the list of all the species the proteins matching this entry are found in.
Taxonomy tree of all the species the proteins matching this entry are found in.
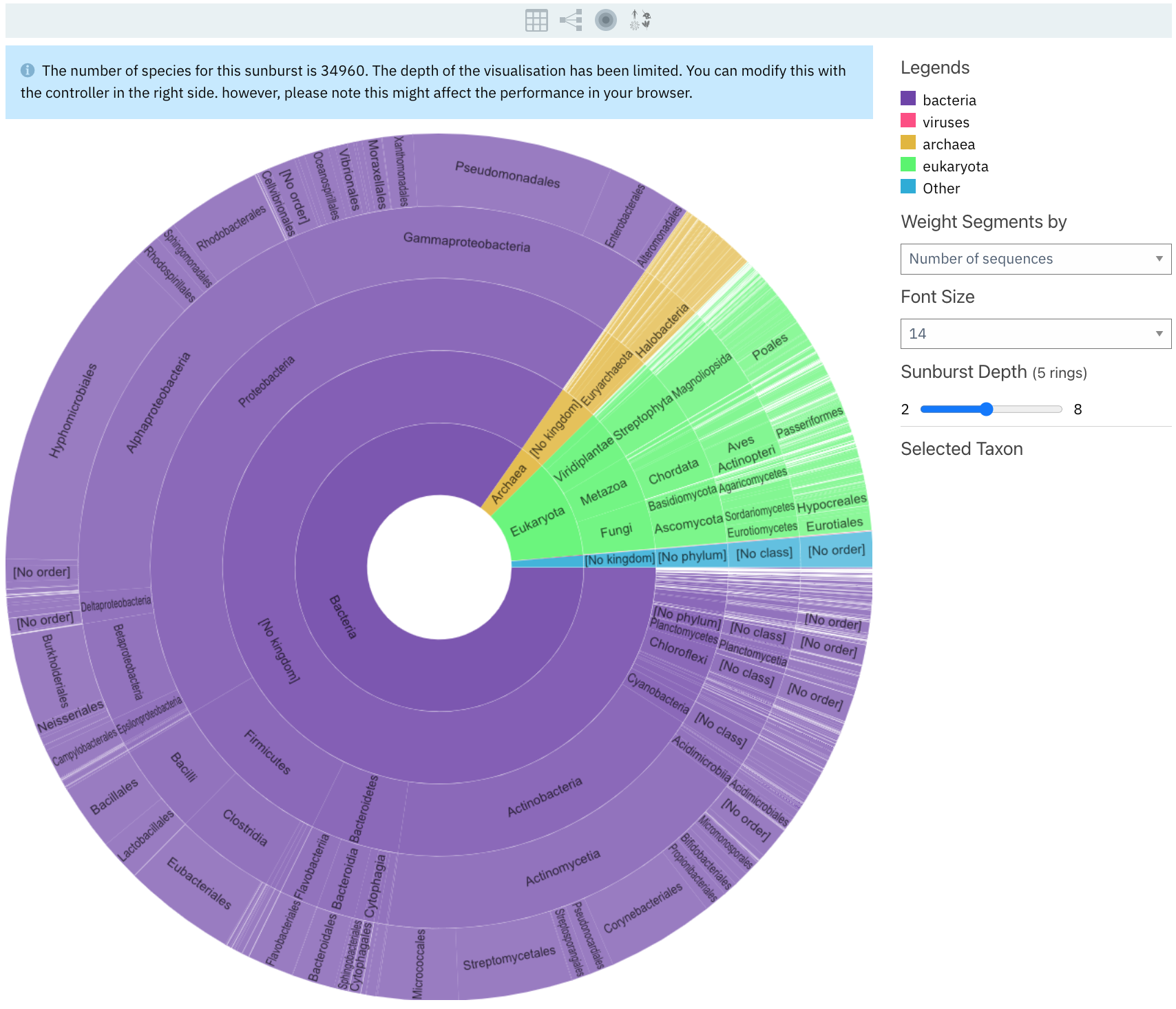
Sunburst view displays the taxonomy distribution of the proteins matching the entry, from the least specific at the centre to more specific going towards the outside.
Table with the number of proteins found for key species, these are 12 model organisms commonly used in scientific research: Oryza sativa subsp. japonica, Arabidopsis thaliana, Homo sapiens, Danio rerio, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Escherichia coli, Escherichia virus T4, Halobacterium salinarum.
Sunburst is the default view of the subpage. A range of options can be selected to customise the view:
The segment size can be adjusted based on the number of sequences matching a taxon (default) or by the number of species per taxon.
The sunburst depth can be adjusted between 2 to 8 rings.
In the table views, for each organism, the taxonomy identifier and protein count information are provided. The ACTIONS column offers the possibility to:
View all the protein matches in the Proteins tab
Download a FASTA file of the protein matches
View the taxonomy information in the Taxonomy entry page
If the first option is selected, a table with all the corresponding proteins is displayed. For each protein, we can see the UniProt ID, name, corresponding gene, the organism where it is found, a link to the protein structure prediction (AlphaFold) and a small protein viewer that highlights the region of the protein matched by the InterPro entry.
Proteomes¶
List of proteomes whose members are represented by proteins matching this entry. A proteome represents a set of proteins whose genomes have been fully sequenced. A given taxonomy node may have one or more proteomes, for example, to reflect different assemblies of a genome. Proteome data is imported from UniProt proteomes. For each proteome, the same set of actions are available than the ones in Taxonomy, the taxonomy information being replaced by proteome information in the Proteome entry page.
Structures¶
List of structures from the PDBe database that match to protein sequences included in this entry.
AlphaFold¶
AlphaFold protein structure predictions are generated by DeepMind [4]. AlphaFoldDB now includes the Big Fantastic Virus Database (BFVD) protein structure predictions, which are generated by using ColabFold applied to the viral sequence representatives of the UniRef30 clusters.
At the top of the page a 3D viewer (powered by Mol*) shows an interactive view of the predicted structure for one of the proteins matching the InterPro entry. The structure is coloured by per-residue plDDT score, it can be zoomed in and out, and rotated. Clicking on a residue induces a zoom in effect and displays contacts with surrounding residues, clicking on the blank area around the structure zooms out.
The protein accession and organism are displayed on the left hand side, together with links to the corresponding InterPro protein page and the AlphaFold and Foldseek Server websites. The model confidence colour scale, determined using the plDDT score, is also displayed, varying from dark blue (very high confidence) to orange (very low confidence). Immediately below the protein structure viewer, a table shows the available multimer predictions (monomers -default-, homodimers and heterodimers) that can be chosen for display.
The data can be downloaded in PDB or mmCIF format, by clicking on the corresponding buttons below the 3D viewer.

AlphaFold structure predictions tab for IPR000562, UniProt O60449.¶
On an InterPro entry page, below the 3D viewer, a table containing the list of UniProt accessions matching the InterPro entry for which structure predictions have been generated is shown. For each protein it is possible to:
Access the Protein entry page by clicking on the UniProt accession or name
Access the Taxonomy entry page by clicking on the species
Display the structure prediction on the current page by clicking on the Show prediction button.
On a the AlphaFold tab of a protein entry page, below the 3D viewer, the protein sequence viewer displays the member database signatures and InterPro entries matching the protein. Hovering over a match highlights the corresponding section in the predicted structure 3D view. Besides, on top of the protein structure viewer there is a drop-down list that allows the user to colour the structure according to the per-residue plDDT score or AlphaFold model confidence (default), the predicted TED domains, the representative families or the representative domains (if available).

Pathways¶
List of pathways identified for protein sequences included in this entry. This information is provided by the MetaCyc Metabolic Pathway Database and the Reactome database.
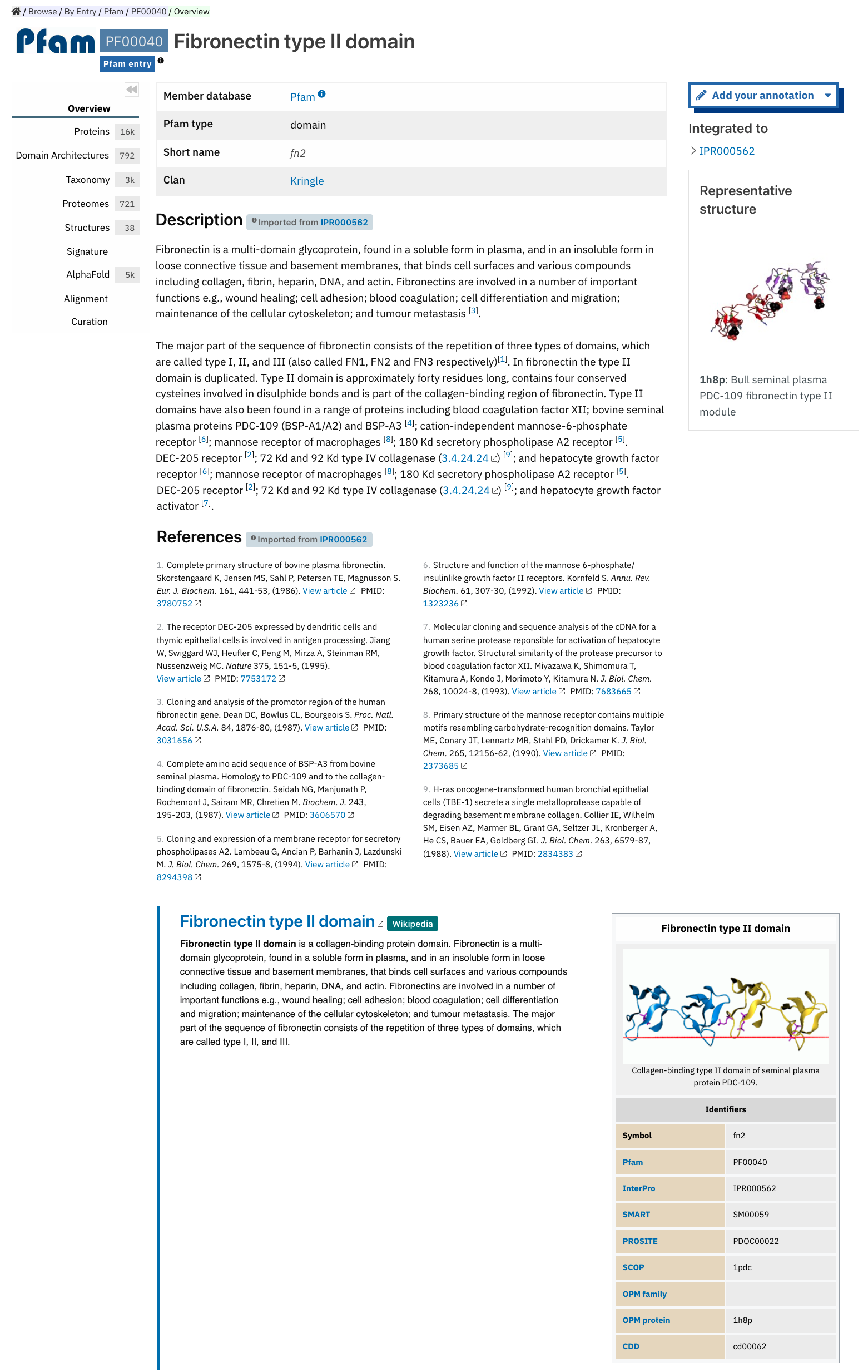
Member database page¶
InterPro provides entry pages for each signature that a member database holds. This includes signatures that have not yet been, or can’t be, integrated into InterPro (unintegrated signatures).

Member database signature entries provide information about which database the signature is from, the signature accession, the type of entry as defined by the member database (e.g. family, domain or site), and the short name given to the entry by the member database.
Some member databases provide a description giving information about the family/domain or site function, when this is not the case and the signature is integrated in an InterPro entry, the InterPro description is displayed.

InterPro member database page for CATH-Gene3D signature G3DSA:1.10.10.10.¶
To address the absence of annotations for certain member database signatures that are not integrated into any InterPro entry, we’ve employed AI to automatically generate descriptions by extracting information from Swiss-Prot. It’s important to note that these descriptions have not undergone curator review, and we advise regarding them as preliminary sources of information. Provide feedback links to a Feedback page, which allows the user to suggest updates to the annotation. Read more about AI-generated content.

InterPro member database page for PANTHER signature PTHR13944.
AI-generated content is accordingly flagged with an tag.¶
Some member databases create groups of families that are evolutionary related. Pfam calls them clans, CDD uses the term superfamily and, for PIRSF and Panther the concept is associated with the parent families of their hierarchy. We use the umbrella term Clan to refer to Pfam groups and Set to refer to the other groups. When available, the set/clan to which the signature belongs to is indicated.
The right hand side of the page provides links to the InterPro entry in which this signature has been integrated, and an external link to the signature on the member database’s website when available. At the bottom of this column, if any experimentally solved structure is available, a Representative structure shows a small static 3D representation, the corresponding PDB ID and name and a link to the structure entry page. For Pfam signatures, the Provide feedback button links to a Feedback page, which allows the user to suggest updates to the Pfam annotation.
For signatures provided by the Pfam member database, a short extract of one or more Wikipedia pages are also displayed when available to complete the description.
In addition to the Proteins, Taxonomy, Proteomes and Structures tabs, member database pages may also display information in the following additional tabs: Domain architectures, AlphaFold, Profile HMM and Alignment.
Profile HMM¶
Profile HMM information are available for the NCBIfam, Pfam, PANTHER, PIRSF and SFLD member databases. At the top of the page, general HMM information about the HMM are displayed, including the HMM build commands and Threshold used, and offers the possibility to download the HMM profile defining the signature.
The profile HMM representing the model that defines the entry is visualised in this page as a logo, using Skylign. The visualisation displays the amino acid conservation for each residue in the model. To navigate large logos, the user can drag the rendered area to a desired position. Alternatively, the user can input a residue number to be viewed. When selecting a particular residue in the logo, the probabilities of each amino acid are displayed in the bottom part.

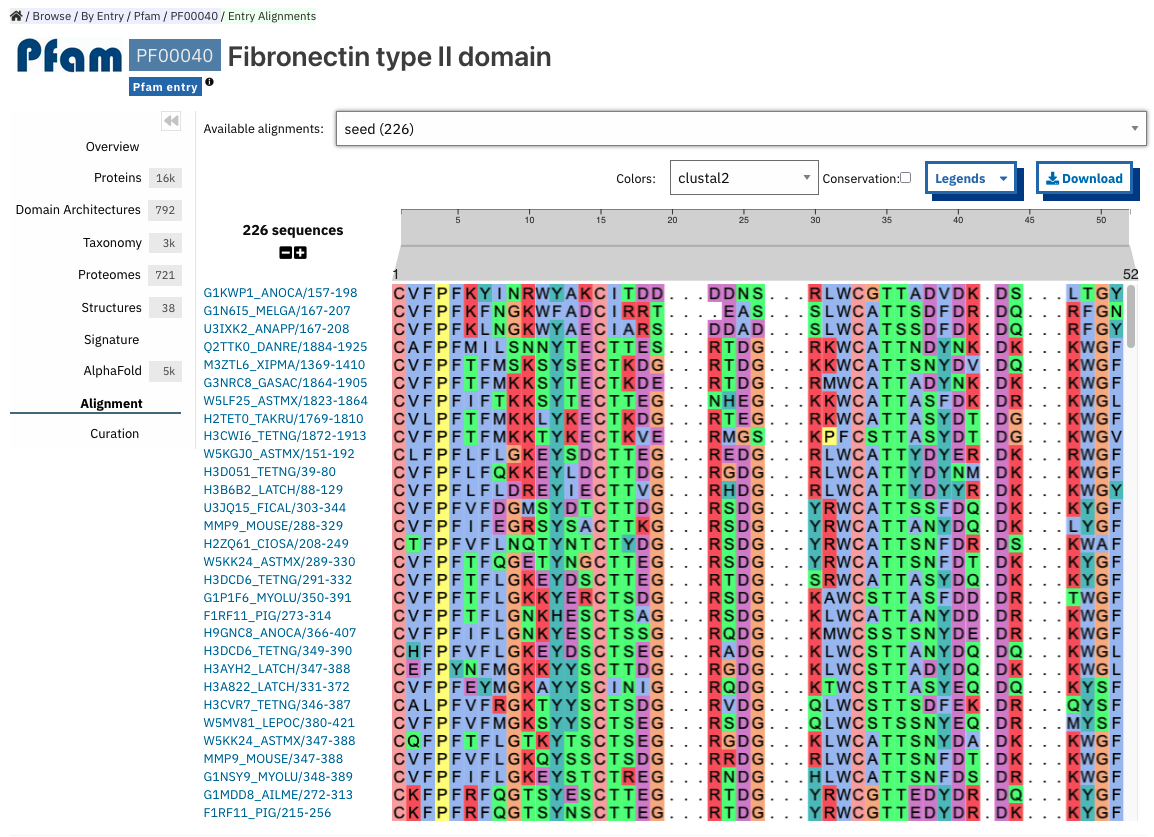
Alignment¶
This section allows users to view and download any available alignment file that is associated with the current member database signature. Currently, the alignment files are only available for the Pfam member database, but hopefully we will be able to include alignments for other member databases in the future.
First, one of the available alignments has to be selected. For example in the image below the user has selected the “seed” alignment. If the selected alignment has more than 1000 sequences, a warning message appears to inform users that big alignments can cause memory issues in the browser. A compressed file (gzip) of the current alignment is available by clicking on the Download button.
Interacting with the grey navigation bar over the sequences allows users to navigate the alignment; dragging the left and right limits of the navigation bar allows users to zoom to a particular position or adjust the zoom level. Alternatively, the zoom level can also be defined by scrolling up/down while holding the [ctrl] key. Scrolling up/down allows to move other sequences in the alignment into the visible area of the viewer.
Subfamilies¶
This section provides a list of subfamilies derived from the signature and a link to get more information in the member database website. Currently, this list is available for the PANTHER and CATH-Gene3D member databases. For PANTHER subfamilies, the GO terms associated to them are also displayed.
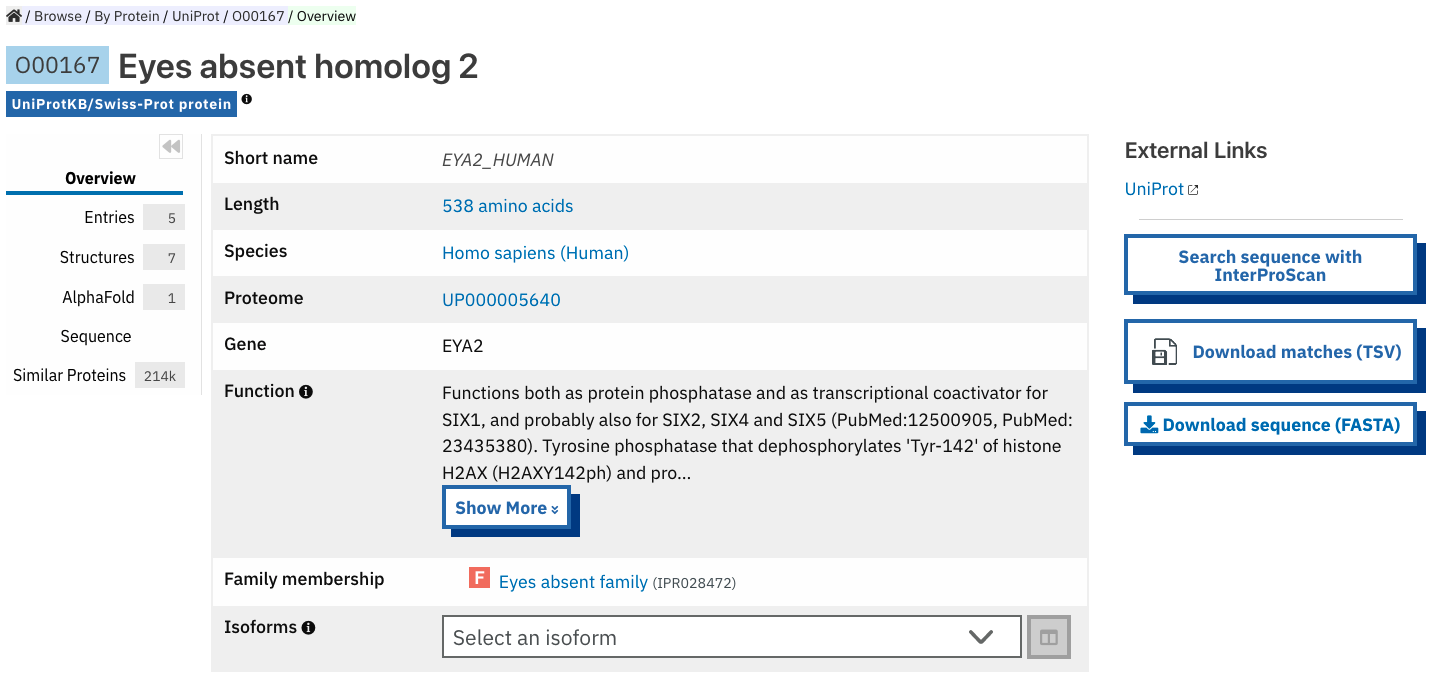
Protein entry page¶
The Protein entry page contains information on a specific protein provided by UniProt. Protein pages can be accessed either by entering a UniProt accession or identifier in a Text search or by clicking on a protein accession from the Proteins tab in an entry page.
The protein page provides the protein accession, the short name (identifier) given to the protein by Uniprot, the length of the protein sequence, species in which the protein is found, the proteome it belongs to, the gene encoding for the protein and a brief description of the protein’s function where known. All the InterPro family entries this protein is matching are listed under “Protein family membership”. On the right hand side of the page, external links to the protein entry in the Uniprot and/or AlphaFold websites (depending on availability); and the possibility to start a search in Foldseek, perform an InterProScan search for the full length or part of the protein sequence, download the sequence in FASTA fomat and export the matches in TSV format are provided.
The protein entry page also displays the protein sequence viewer to show the associated domains, sites etc.
When available, GO terms associated to InterPro entries and PANTHER families are displayed at the bottom of the page. GO terms provide information about Biological processes, Molecular function and Cellular components.
The following tabs may be available: Entries, Isoforms,:ref:structures, Sequence, Similar proteins and AlphaFold.
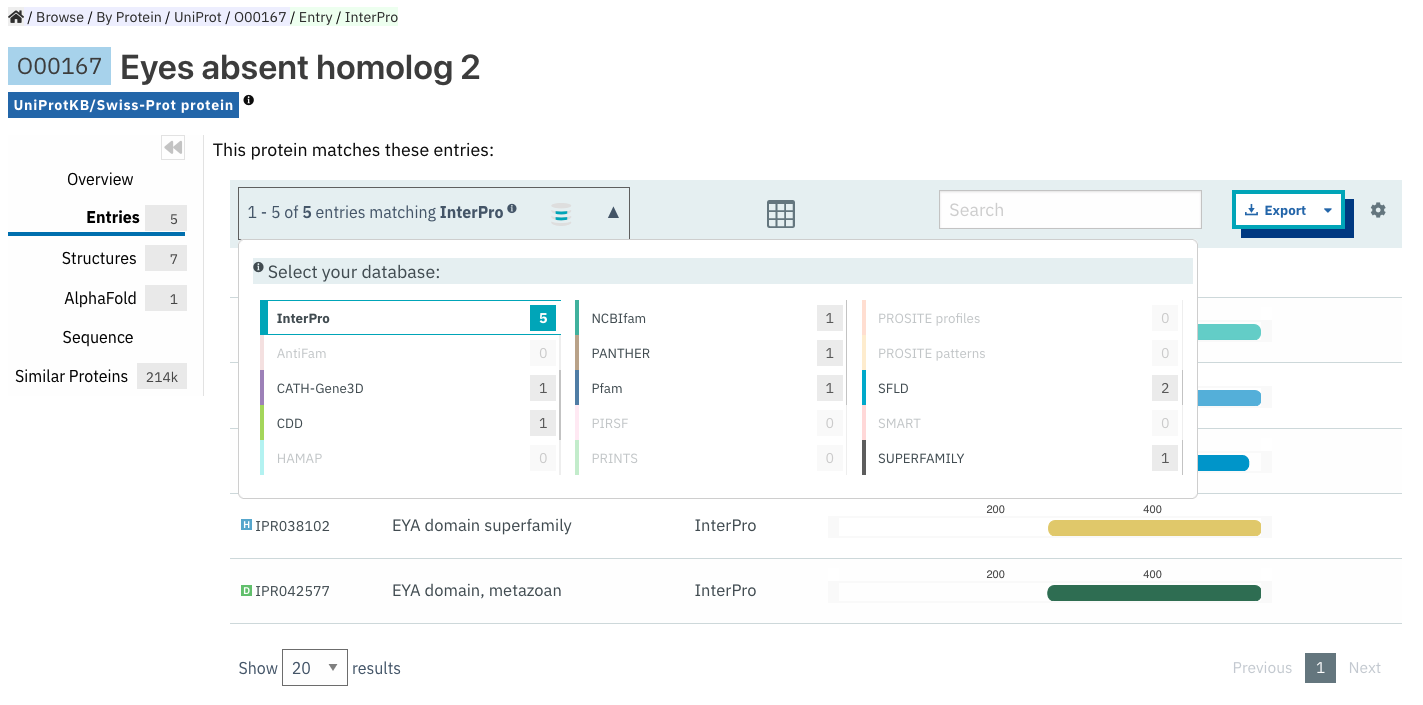
Entries¶
List of InterPro entries that include this entity. The results can be filtered by member databases using the dropdown box located on the left side of the header of the result table. This functionality is available for all the tables presenting InterPro entries in the website.
Isoforms¶
When available, different isoforms of the protein can be selected to compare their structural prediction and InterPro matches with the consensus protein sequence. When an isoform is selected, a new protein sequence viewer corresponding to the selection is displayed, with the corresponding structural prediction coloured track. The link available by clicking ‘pLDDT’ in the protein sequence viewer leads to the structural prediction of the specific isoform in the AlphaFold website.
Sequence¶
This tab shows the protein FASTA sequence. The full sequence or part of the sequence (by selecting the region of interest) can be used to perform an InterProScan search.
Similar proteins¶
List of proteins that have the same domain architecture as this protein, including the Pfam/InterPro accession for each domain. The list can be filtered to either show all the protein matches or only the reviewed proteins from UniProt. For each protein the UniProt ID, name, length, corresponding gene, the organism where it is found and a link to the protein structure prediction page (AlphaFold).
UniParc page¶
When a specific protein is not included in UniProt, the corresponding UniParc sequence entry with its protein sequence viewer will be displayed. A link to the entry’s history in UniProt is provided at the top right side.
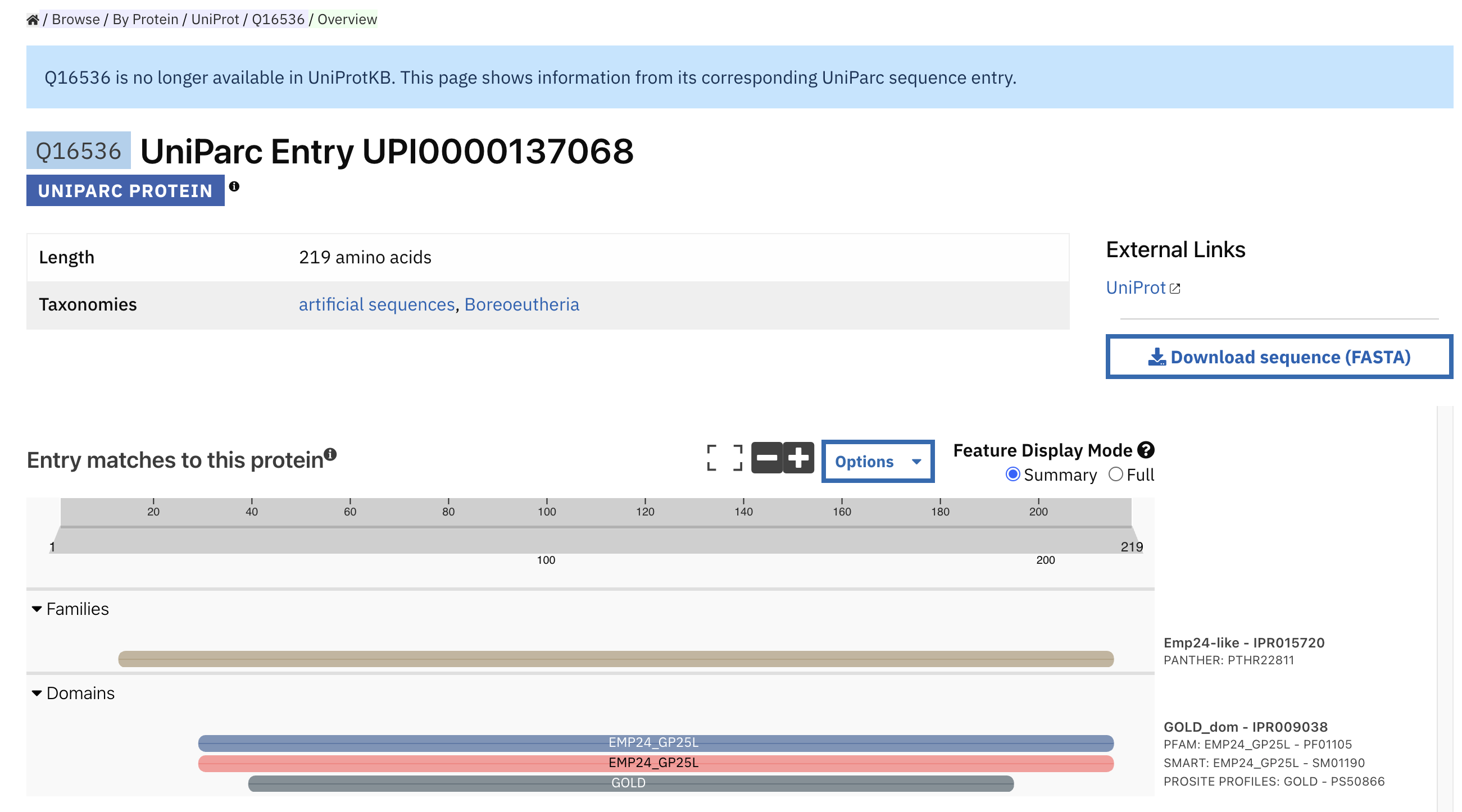
Uniparc page for the protein sequence Q16536, corresponding to UniParc entry UPI0000137068.¶
Structure entry page¶
InterPro provides entries for all the structures available in the Protein Data Bank in Europe (PDBe). A structure search can be performed by clicking on a structure provided in a results list or by entering the protein structure accession in the Quick search box (magnifying glass symbol) or by performing a Text search.
At the top of the structure page, general information about the structure is displayed: the structure’s accession number (PDB ID), resolution, release date, the method used to determine the structure (e.g. “Xray”) and the chains composing the structure. External links to PDBe, RCSB PDB, PDBsum, CATH, SCOP, ECOD, Proteopedia and Foldseek are provided on the right hand side of the page.
Following, the general information section, a 3D viewer (powered by Mol*) shows an interactive view of the 3D structure. Hovering over a residue displays the name of the entry, the chain and residue information below the viewer. Clicking on a residue in the viewer induces a zoom in effect and displays contacts with surrounding residues, clicking on the blank area around the structure zooms out. Below it, the protein sequence viewer with the InterPro matches is displayed for each unique chain (to avoid redundancy, if a structure contain several chains that are identical copies of the same protein, only one protein sequence viewer will be displayed for this specific protein). This protein viewer has an extra category representing the secondary structure information. Hovering over one of the tracks highlights the corresponding region of the protein structure in the 3D structure viewer.
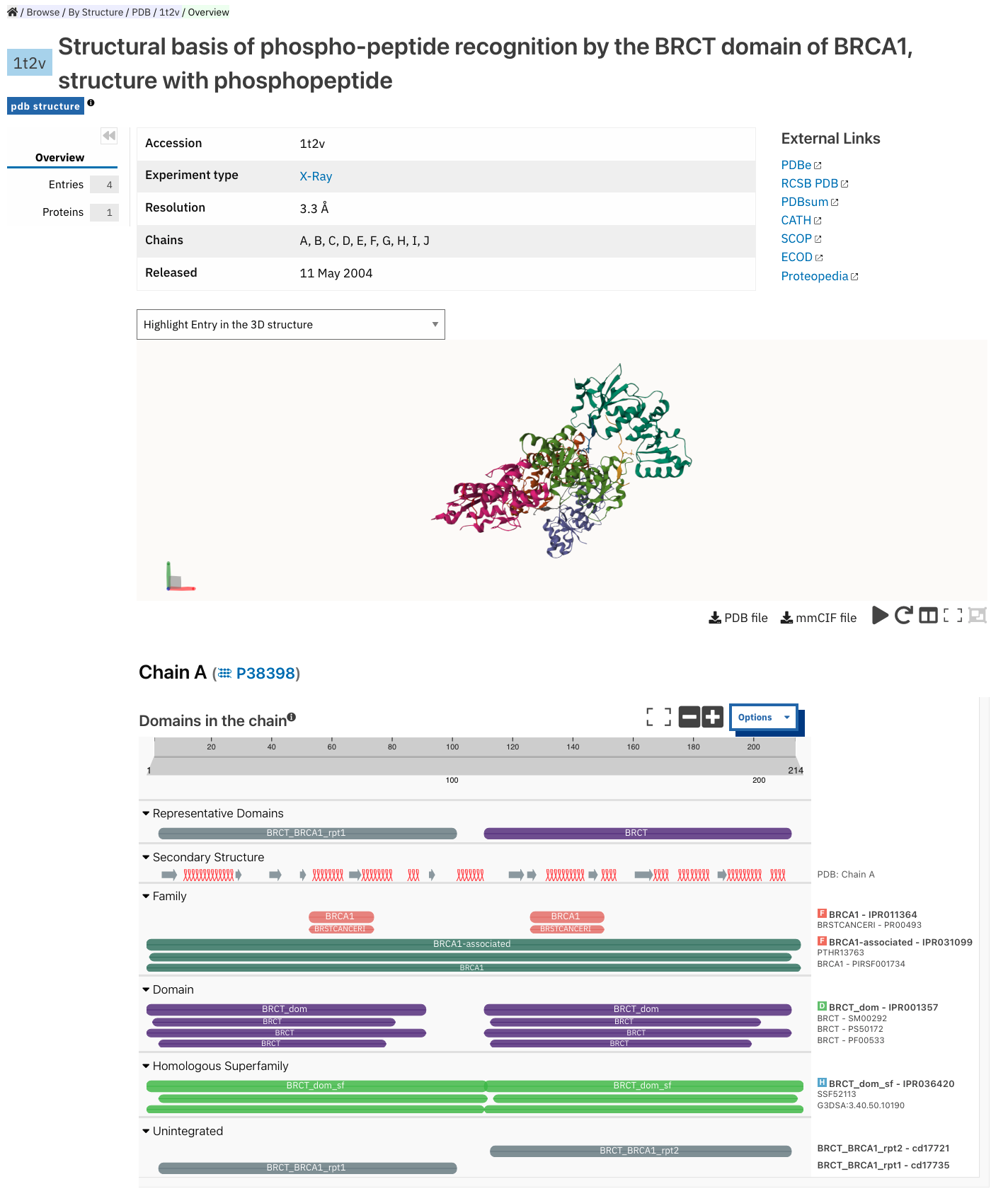
More information is available on the corresponding train online section.
Taxonomy entry page¶
Taxonomy pages display the name, taxonomy ID, lineage and children nodes for a particular taxon. Any reference to this taxon from another page throughout the website will link to this page.
The overview also includes a graphical representation of the lineage of the selected taxon. The nodes in the visualisation are also links, so you can jump to the page of a particular taxon of interest.
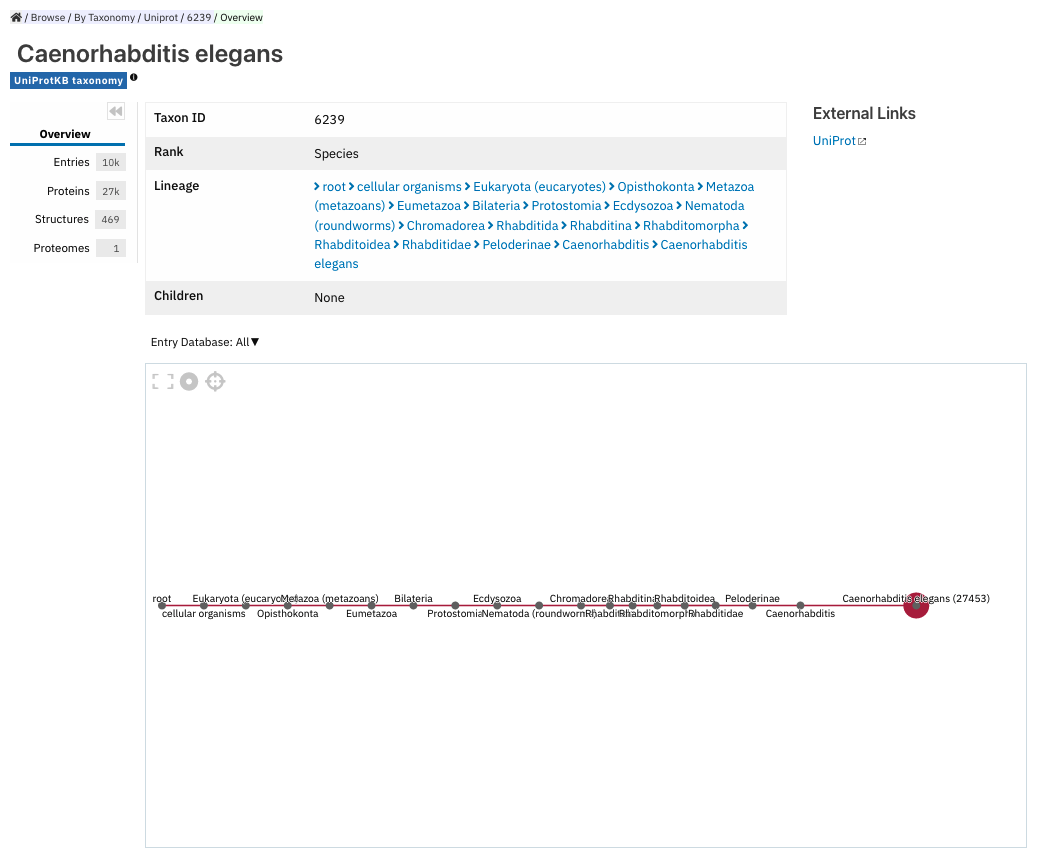
Taxonomy entry page for Caenorhabditis elegans.¶
The following tabs may be available: Entries, Proteins, Structures and Proteomes.
Proteome entry page¶
The proteome entry page displays general information provided by UniProt: its ID, strain, and a description of the organism. It also provides a link to the corresponding taxonomy page.
On the right-hand side, external links to the proteome page in UniProt and the genome page in Rfam are provided, when available.
The following tabs may be available: Entries, Proteins and Structures.

Proteome entry page for UP000001940.¶
When clicking on the Entries tab, the list of InterPro entries matching any sequence in the proteome is displayed. By clicking on the dropdown menu in the table header, the list of entries from a member database can be displayed instead by selecting the database of interest.
Set/Clan entry page¶
Some InterPro member databases create groups of families that are evolutionary related, called sets/clans. This page offers an overview of a specific set/clan provided by a member database, it includes a short description and an interactive view of the signatures included in the set/clan. For the interactive view, different label types can be chosen through the Label Content menu: Accession, Name and Short name. For clans provided by the Pfam member database, an additional section provides literature references and/or Wikipedia articles, when available.
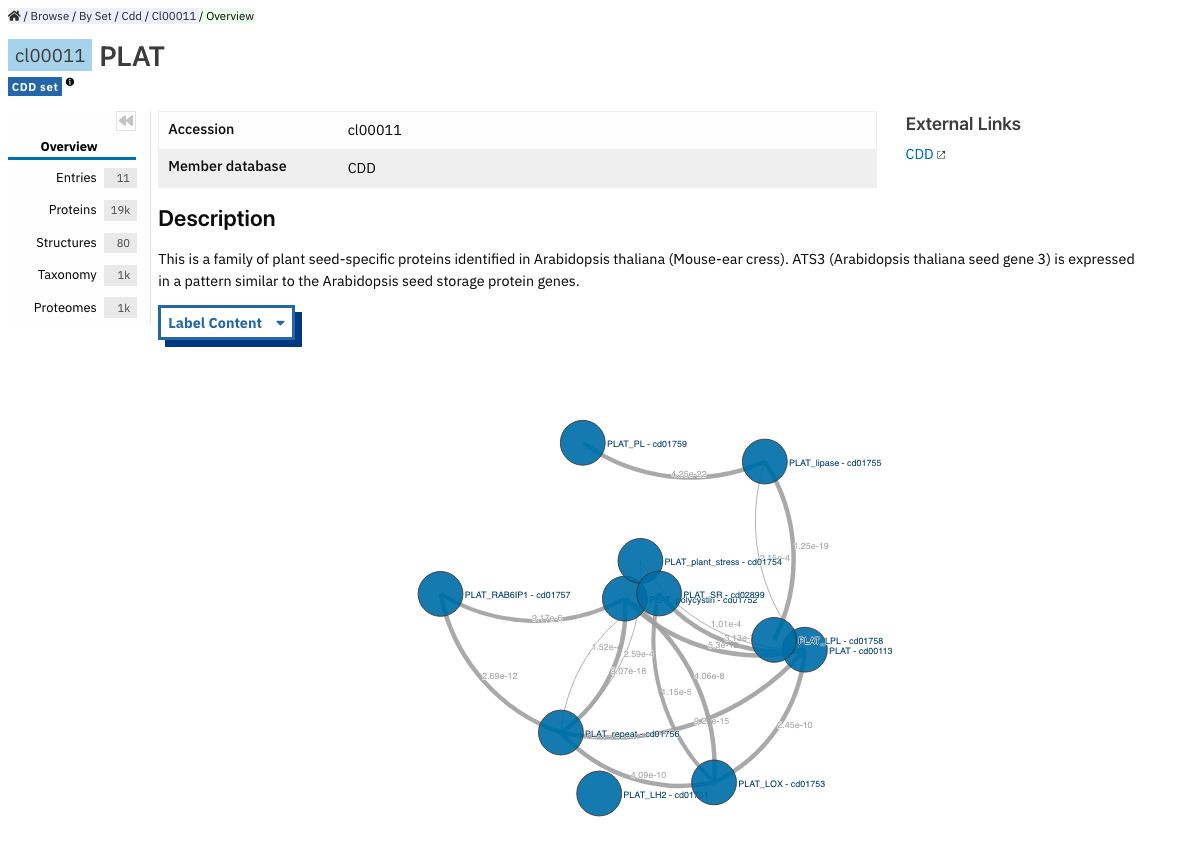
The following tabs may be available: Entries, Proteins, Structures, Taxonomy and Proteomes.
Entries¶
Provides the list of signatures included in the set/clan (accession, name and short name).
For Pfam clans, the Entries tab contains the list of Pfam entries included in the clan and links to the entries SEED alignment and domain architectures pages.